pipeline-resources
About MGscan Metagenomics pipeline
This pipeline is designed to conduct taxonomy profiling and diversity analysis on metagenome shotgun sequencing data. Please find the sample report here.
Source of the pipeline
This pipeline is originally adapted from community-developed nf-core/taxprofiler pipeline version 1.0.0. Zymo Research made significant contributions in the adaption effort. This mainly include the addition of downstream analyses such as diversity analysis, addition of sourmash as the profiler, addition of antimicrobial resistance gene identification and the improvement of the report.
What is in the pipeline
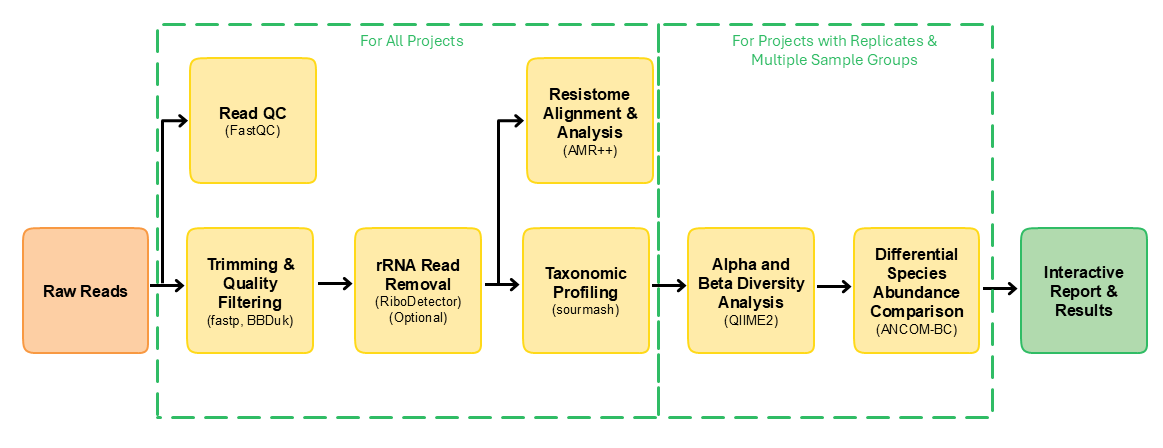
This pipeline is built using Nextflow. A brief summary of pipeline:
- Read QC (
FastQCorfalcoas an alternative option) - Performs optional read pre-processing (code for long-read inherited from nf-core/taxprofiler, but not separately tested by us yet)
- Perform Host-read removal
- Run merging when applicable
- Identifies antimicrobial resistance genes in samples from database MEGARes version 3
- Reads are aligned to MEGARes reference (
bwa mem) - Resistome statistics are quantified and compiled for each sample (
AMRplusplus)
- Reads are aligned to MEGARes reference (
- Performs taxonomic profiling using one of: (nf-core/taxprofiler has more choices for this step, if there are tools you’d like for this step, please let us know.)
- Merge all taxonomic profiling results into one table (
Qiime2) and generate composition plot (Krona) - Perform alpha/beta diversity analysis (
Qiime2) and beta diversity differential expression (ANCOM-BC) - Present all results in above steps in a report (
MultiQC)
For details, please find the source code here.
Default pipeline parameters
Trimming (fastp)
fastp automatically detects and trims adapter sequences. Reads shorter than 15bp after trimming are discarded.
Low complexity filtering (BBDuk)
BBDuk discards any read that has less than 0.3 in average entropy in a sliding window of 50bp.
Taxonomic classification (sourmash)
sourmash sketch is run with the parameter of k=31,scaled=1000,abund. sourmash gather is run with the parameter of --ksize 31 --threshold-bp 5000. This means that k-mers of length 31 are profiled at 1:1000 downsampling with abundance of each k-mer consdiered. A match/genome will only be reported if the estimated overlap between a sample and the reference genome is at least 5000bp.
Sourmash database
By default, the database option sourmash-zymo-2024 include the following reference genomes.
- GTDB R08-RS214 genomic representatives
- Genbank viral genomes(2022)
- Genbank archael genomes(2022)
- Genbank protozoa genomes(2022), with one genome GCA_002893375.1 removed due to contamination
- Genbank fungi genomes(2022)
- A list of 29 genomes realted to ZymoBIOMICS Microbial Community Standards
- A list of 16 common host genomes such as human, mouse, rat, etc. These genomes are included for host reads removal purposes. Their reads are not included downstream analysis.
K-mer to read approximation
We estimate the numbers of reads belonging to each microbial genome by multiplying f_unique_weighted values of that genome from sourmash to the total number of reads after trimming and low complexity filtering.
Sample filtering criteria
The following samples not removed from downstream analysis at various steps:
- Samples with a file size <1 kb after trimming and read length filtering
- Samples with no identified microbial genomes by sourmash after host genome removal
- Samples with less than 1,000 total reads assigned to non-host genomes
- Samples that fail to produce a sourmash result after 3 tries with increased compute resources, up to 180GB of memory. This rarely happens and only happens to samples with extremely complex profile.