pipeline-resources
About MGscan Metatranscriptome pipeline
This pipeline is designed to conduct de novo metatranscriptome assembly and taxonomic and functional annotations of the assembly from NGS metatranscriptome data. Please find the sample report here.
Source of the pipeline
This pipeline was adapted from community-developed nf-core/metadenovo pipeline version 1.0.0. Zymo Research made significant contributions in the adaption effort. This mainly include adding a removal of rRNA reads step, development and use of custom version of CAT, taxonomy plots and the improvement of the report.
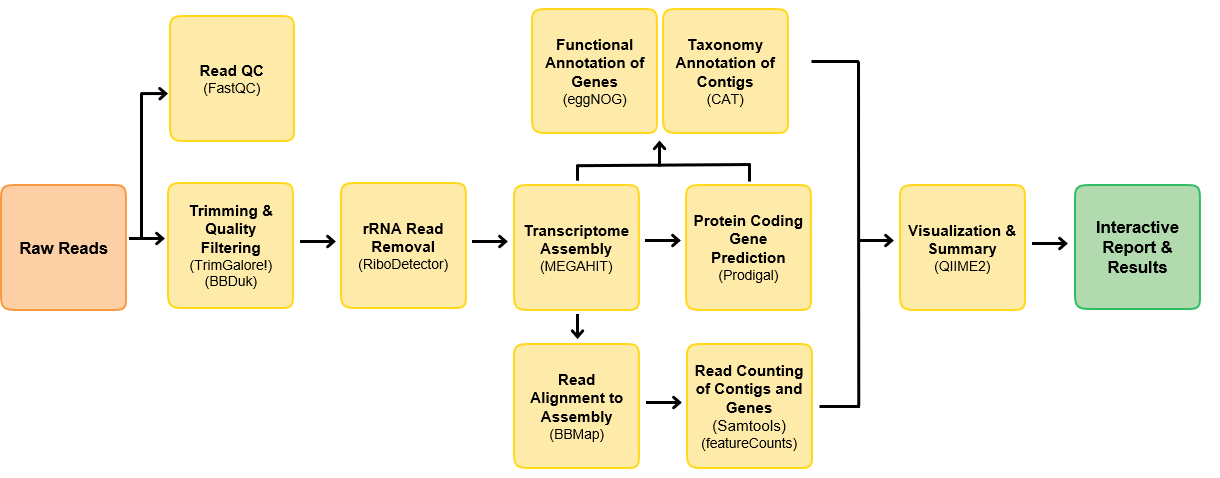
What is in the pipeline
This pipeline is built using Nextflow. A brief summary of pipeline:
- Read QC with
FastQC - Performs read pre-processing
- Adapter clipping trimming with
TrimGalore - Low complexity and quality filtering with
bbduk
- Adapter clipping trimming with
- rRNA reads removal with
RiboDetector - de novo assembly with either
MEGAHITorRNAspades - Align reads back to assembly with
BBmap - Get read counts per contig with
Samtoolsand per gene withFeaturecounts - Gene prediction with
Prodigal - Taxonomic annotation of contigs with a custom version of
CAT - Functional annotation of genes with
eggNOG mapper - Merge taxonomic and read count results, draw composition bar plots with Qiime2
- Present results in a report with
MultiQC
For details, please find the source code here.
Default pipeline parameters
Low complexity filtering (BBDuk)
BBDuk discards any read that has less than 0.3 in average entropy in a sliding window of 50bp.
rRNA reads removal (RiboDetector)
The CPU version of RiboDetector is run with the average read lengths dynamically calculated before input. The rRNA classification method for paired end reads is set as none.
Assembly (Megahit)
The default assembler is MEGAHIT with all its default settings. The minimum contig length is 200bp.
Align reads to assembly (BBmap)
The reads are aligned back to assembly using BBmap with the parameters minid=0.9 idfilter=0.9 maxindel=20. This means minimum sequence identity of 90% and maximum insertion or deletion of 20bp.
Gene prediction (Prodigal)
Gene prediction on contigs was run using prodigal with -p meta mode.
Taxonomy annotation (CAT)
Taxonomy annotation was done using a customized version of CAT found here. In short, the custom version compensates for seqeuences with bad taxonomies in the NCBI NR database. The database used, 20231120_CAT_nr was downloaded from CAT directly. The parameter used here are -r 10 -f 0.5 --orf_support 0.9 --ignore_notax_hits. Note that -r 10 -f 0.5 are parameters from the original version of CAT. The other two parameters are added in the custom version. Please find details in its GitHub repo.
Functional annotation (eggNOG)
The database for eggNOG-mapper was downloaded using eggNOG’s download_eggnog_data.py on May 2024.
All other steps are using the software’s default settings.