pipeline-resources
About MGscan 16S/18S/ITS pipeline
This pipeline is designed to process microbial amplicon sequencing data, performing taxonomic assignment of the prokaryote 16S ribosomal RNA gene, the eukaryote 18S ribosomal RNA gene and the ITS (internal transcribed spacer). Supported data types include single- or paired-end Illumina and PacBio reads.
Source of the pipeline
This pipeline was originally adapted from community-developed nf-core/ampliseq pipeline version 2.4.0.
What is in this pipeline
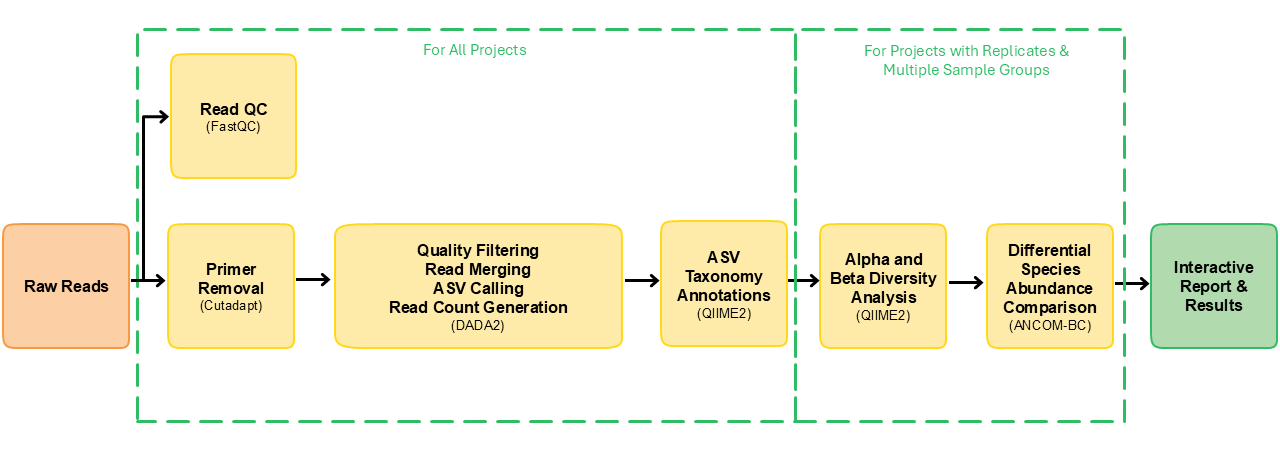
This pipeline was built using Nextflow. A brief summary of pipeline:
- Raw read QC (
FastQC) - Read trimming using (
Cutadapt) - Infer Amplicon Sequence Variants (ASVs) (
DADA2) - Taxonomic classification using
QIIME2 - Excludes unwanted taxa, produces absolute and relative feature/taxa count tables and plots, plots alpha rarefaction curves, computes alpha and beta diversity indices and plots thereof (
QIIME2) - Calls differentially abundant taxa (
ANCOM-BC) - Presents all QC and analysis results in a comprehensive report (
MultiQC)
For details, please find the source code here.